
Review / Open Access
DOI: 10.31488/jjm.1000127
Pathogenesis of Hypervirulent Group A Streptococcus
Mengyao Liu, Benfang Lei*
Department of Microbiology and Immunology, Montana State University, Bozeman, MT 59718, USA
*Corresponding author:Benfang Lei,Department of Microbiology and Immunology, Montana State University, Bozeman, MT 59718, USA
Abstract
Group AStreptococcus(GAS) causes common pharyngitis and skin infections and occasional severe invasive infections. This review describes the recent progress on the pathogenesis of hypervirulent GAS.CovRSmutations are frequent among invasive GAS isolates and lead to hypervirulence. GAS CovRS mutants can be selectedin vivo by neutrophils. The role of protease SpeB in source-sink dynamics of wild-type GAS and hypervirulent variants is discussed. Streptolysin S and PAF acetylhydrolaseSse critically and synergistically contribute to the inhibition of neutrophil recruitment by GAS CovS mutants. CovS mutations in emm3 GAS lead to the vascular invasion and enhance systemic GAS dissemination.
Keywords: Group A Streptococcus, Streptococcus pyogenes, necrotizing fasciitis, pneumonia, vascular invasion, perivascular interstitium, virulence, mutation, CovRS, Sse, PAF acetylhydrolase, Protease, SpeB
Introduction
Streptococcus pyogenes, which is commonly referred asGroup AStreptococcus (GAS), is a major human pathogen that causes both relatively mild common infections, such as pharyngitis and superficial skin infections, and potentially lethal, severe invasive infections, including soft-tissue infections, pneumonia, and necrotizing fasciitis [1, 2]. These invasive infections often lead to systemic GAS dissemination, resulting in septic shock and streptococcal toxic shock syndrome. Progress has recently made on pathogenesis in severe invasive GAS infections that include the understanding of the role of CovRS mutations in hypervirulence, in vivo selection of GAS CovRS mutants, and innate immune evasion and vascular invasion by hypervirulent GAS CovRS mutants. This mini review focuses on the progress in these areas. Because of the limited scope, we do not intend to provide a thorough review on the subject butemphasize more on our own contributions.
Hypervirulent Group AStreptococcus Variants
Severe GAS infections were frequent and often fatal in the 19th century and reemerged in the 1980s. The reemergence of severe invasive GAS infections in the 1980s is associated with the emergence of the virulent M1T1 clone of genotype emm1 GAS and virulent emm3 GAS. The M1T1 clone of emm1 GAS has been evolved by the acquisition of DNase Sda1- and superantigenSpeA-encoding prophages and the replacement of a 36-kb chromosomal region of pre-1980 emm1 GAS with that of emm12 GAS that contains the NADaseand streptolysin O genes [3]. Contemporary M3 GAS acquired a prophage that encodes the superantigenSpeK and phospholipase A2 SlaA[4]. Since 2000, M89 GAS with the loss of the genes for synthesis of the hyaluronic acid capsule has also emerged to cause severe invasive infections[5]. The 5 most prevalent emm genotypes of recent pharyngeal and invasive GAS isolates are emm1 (M1T1 clone), emm3, emm12, emm28, and emm89 [2, 6]. Invasive emm3 GAS causes a higher mortality rate than invasive strains of other genotypes [7].
Invasive GAS isolates are usually more virulent than pharyngeal isolates in experimental animal infections, displaying greater tissue invasion, innate immune evasion, and systemic dissemination than pharyngeal isolates[8, 9]. Invasive GAS isolates frequently carry mutations of the two-component regulator systemCsrRS or CovRS[10, 11].CovRS negatively regulates many virulence factors, including many of those that are involved in the evasion of innate immunity [12]. Natural CovRSmutationsenhance virulence gene expression, innate immune evasion, systemic dissemination, leading to hypervirulence[8, 9, 12-14]. Natural nonsense mutations of the orphan kinase RocA in emm3 and emm18 GAS also contribute to enhanced virulence[15, 16]. Invasive emm3 GAS isolates also have prevalent mutations in RopB, the activator that is essential for the expression of the protease SpeB[10, 11].
In vivo Selection of Hypervirulent GAS Mutants
The emergence of CovS mutations during infection has been demonstrated in M1T1 and emm12 GAS during experimental mouse infections, and CovS mutants have higher expression of virulence genes and virulence [6, 13]. In vivo selected CovS mutants of M1T1 GAS has downregulated expression of SpeB, lacking detectable levels of the SpeB activity in culture supernatant (SpeB activity-negative or SpeBA-phenotype) [14]. The SpeBA-phenotype is a validated marker for selected CovS mutants of CovS of M1T1 and M12 GAS in mouse infection[6, 20].
No SpeBA- variants can be detected after cutaneous infection with SpeBA+emm3 isolates, and the emergence of emm3 GAS CovS mutants cannot be demonstratedin mice infection by screening for variants with the SpeBA-phenotype[6]. These results are surprising becauseCovS mutations are frequent in clinical invasive emm3 isolates [10, 11]. However, the failure to demonstrate the emergence of emm3 GAS CovS mutants in mouse infection may be just a technical difficulty that arises in a possible distinction between emm1 and emm3 GAS in regulation of speB. The natural CovSG457V point mutation of invasive emm3 isolate MGAS315, like in M1T1 GAS, enhances expression of virulence genes and critically contribute to its virulence; however, this CovSmissense mutation does not cause a SpeBA-phenotype in MGAS315, and CovSG457V mutant cannot confer the SpeBA+ phenotype of covS deletion mutant of M1T1 GAS [9]. Apparently, screening with the SpeB activity assay is unable to identify certain emm3 GAS CovS mutations in mouse infection.
The DNase sda1 has been reported to be required for the in vivo selection of SpeBA- variants of M1T1 GAS, leading to a proposal that Sda1 helps GAS avoid killing by neutrophil extracellular traps in the absence of SpeB production and thereby provides pressure for selection of CovRS mutations with the SpeBA- phenotype [17]. However, the role of Sda1 in the selection of M1T1 GAS SpeBA- variants cannot been confirmed, and deletion of all three DNase genes has no effect on the selection of M1T1 GAS CovS mutantsin mouse infection [18]. The capsule synthase hasA and M protein emm genes are reported to be required for the selection of CovRS mutants of M1T1 GAS [19]. The first sequenced M1 GAS strain SF370, which has functional hasA and emm but does not belong to the M1T1 GAS clone, rarely switch to SpeBA- during experimental mouse infection [18].Thus, the exact switch for in vivo selection of hypervirulentM1T1 GAS CovS mutants remains elusive.
In vivo selection of M1T1 GAS CovS mutants is diminished in neutropenic mice that are obtained by depletion of neutrophils through the treatment of mice with anti-Ly6G monoclonal antibodies 1A8 and RB6-8C5[20]. RB6-8C5 also depletes a subset of monocytes that differentiate into recruited macrophages during infections. However, the role of recruited macrophages in selection of GAS CovS mutants is ruled out based on the phagocyte responses and CovS mutant selection in CXCR2-/- mice. CXCR2 is a receptor for CXC chemokines and is involved in neutrophil recruitment. In vivo selection of M1T1 GAS CovS mutants is significantly reduced in CXCR2-/- mice. CXCR2-/- mice display impaired neutrophil recruitment but have higher recruited macrophages at infection sites in air-sac infection of mice with M1T1 GAS[20]. The decrease in the selection of M1T1 GAS CovS mutants in CXCR2-/-mice is apparently correlated with the impaired neutrophil recruitment but not with the increased recruitment of macrophages. The findings support that neutrophils are required for in vivo selection of GAS CovS mutants.
CovS mutant outcompetes wild-type bacteria in mice with normal neutrophil responses but is outcompeted by wild-type bacteria in neutropenic mice[20]. The finding demonstrates that CovSmutants can survive better than the parent GAS in the presence of neutrophils.
M1T1 GAS mouse infection also selects nonsense and missense mutations of RocA[21]. Selected RocA mutations enhance the expression of many CovRS-controlled virulence genes in vitro and in vivo but, unlike CovS mutations, do not downregulatespeB transcription at stationary growth phase and in subcutaneous infection of mice.
The current model for the occurrence of hypervirulent GAS mutants during infection is presented in Figure1. In this model, spontaneous covS and rocAmutations are selected by neutrophils, and enhance virulence gene expression, enhancing innate immune evasion, tissue invasion, and systemic dissemination and resulting in hypervirulence.
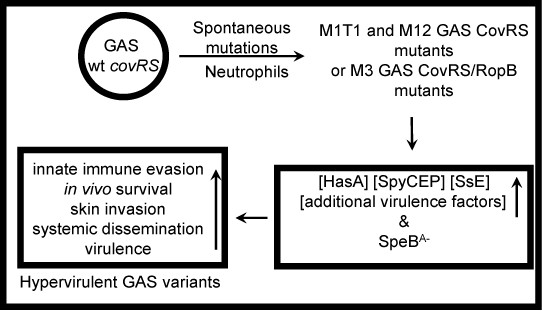
Figure 1.A model for in vivo selection of hypervirulent GAS mutants.M1T1 GAS CovRS mutants. Neutrophils select spontaneous covRS mutations of M1T1 GAS. M1T1 and M12 CovRS mutants and CovRS/RopB mutants enhance expression of multiple virulence genes and downregulateSpeB production, resulting in mutants with enhanced innate evasion, in vivo survival, skin invasion, systemic dissemination, and virulence. The figure was modified from one in reference 20.
Role of SpeBin an Apparent Source-Sink Dynamics of Wild-Type GAS and HypervirulentGAS Variants
Natural M1T1 GAS CovS mutations down-regulate SpeB productionin vitro and in vivo. RopB mutations also down-regulate SpeB expression. M1T1 GAS RocA and CovS mutants have similar enhancement in many CovRS-controlled virulence factors[16] but RocA mutants do not downregulateSpeB productionin vivo[21]. RocA mutants rank between wild type M1T1 GAS and CovS mutants in skin invasion, inhibition of neutrophil recruitment, and virulence in subcutaneous infection of mice[21].RopB deletion of an emm3 isolate, which has a natural RocA mutation, enhances virulence [10]. These findings indicate that, in addition to high expression of CovRS-controlled virulence genes, the downregulation of SpeBis a significant factor for the hypervirulence of M1T1 GAS CovS mutants and emm3CovS/RocA/RopB and RocA/RopB mutants. It has been proposed thatthe downregulation of SpeBpreserves virulence factors to enhance virulence[22]. This notion is supported by the phenotype of covR deletion mutants. Deletions of covR and covS enhance expression of many virulence genes similarly; however, covR deletion enhances SpeB expression.Deletion ofcovRenhances skin invasion but reduces systemic dissemination and virulence[23].SpeB critically contributes to dermal ulceration caused by GAS covR deletion mutant [24]. Apparently, high levels of SpeB expression enhance localized infection and reducessystemic dissemination in soft tissue infections.
The majority of pharyngeal and invasive GAS isolates are SpeBA+[25], suggesting that CovRS mutations arise during human infection with GAS carrying wild-type CovRS and are not transmissible. The source-sink dynamics, a model in ecology that describes how variation in habitat quality affects the population growth or decline of organisms, can be borrowed for a proposal on the relationship of wild-type GAS and its hypervirulent variants and the role of SpeB in this population dynamics (Figure 2). GAS with wild-type CovRS and RopB produces high levels of SpeB, which is critical for causing dermal and mucosal purulent ulceration, degrades virulence factors for localized infection, and may also confer a survival advantage in pharyngeal infection. Thus, SpeB plays a significant role for localized, contagious infection at non-invasive throat and skin surface, which serves as the source habitat of GAS.GAS with CovRS and RopB mutations downregulatesSpeB expression, which preserves virulence factors and indirectly enhances innate immune evasion and tissue and vascular invasion.Thus, the GAS variants cause systemic, less contagious, and potentially lethal infections at usually germ-free tissues and blood, which serves as the sink habitat of GAS.

Figure 2.A proposal for the role of SpeB in a model of source-sink dynamics of wild-type GAS and hypervirulent variants. The high expression of SpeB is critical for localized, contagious infection at the source habitat, non-invasive throat and skin surface whereas the downregulation of SpeB preserve virulence factors to facilitate systemic, less contagious but potentially lethal infections at the sink habitat, the tissues and blood that are usually germ-free.
Evasion of Neutrophil Response by Hypervirulent GAS CovRSMutants
Some necrotizing fasciitis (NF) patients have numerous bacteria but few or no neutrophilic responses at infection sites, which is classified as stage III NF [26], and other histopathologic types include a moderate-to-severe neutrophilic response and a positive Gram staining (stage II) and an intense neutrophilic response with the absence of bacteria (stage I) in infected tissues. Patients with stage III NF have a higher mortality rate than patients with stage I and II NF. Animals in a murine model of NF caused by hypervirulentGAS CovS mutants display stage III histopathologic features of few or no neutrophils at sites of bacterial infection[27, 28].Correction of CovS mutations in hypervirulent GAS CovS mutants enhances the neutrophil response and lead to the Stage II histological type whereas covS deletion of wild-type M1T1 GAS reduces neutrophil recruitment and leads to the Stage II histological typein mouse infection [8,9]. Thus, CovS mutations enhance innate immune evasion.
CovS null mutations enhance the expression of the sse, spyCEP, and scpA genes. Sse, SpyCEP, and ScpA degrade the neutrophil chemoattractant molecules platelet-activating factor (PAF), interleukin 8/CXC chemokine, and C5a peptide, respectively. SsE, but not SpyCEP or ScpA, is required for the inhibition of neutrophil recruitment by hypervirulent M1T1 GAS CovS mutants[8]. Streptolysin S (SLS) delays the exodus of the neutrophils from the vessel lumen into the tissue at the early stage of GAS skin infection in mice [29]. SLS is also cytotoxic to neutrophils [30]. In mouse infection with hypervirulentCovS mutants, Sse and SLS are both required and have synergistic effects for inhibition of neutrophil recruitment and systemic infection.Figure 3 illustrates the critical role of the covS mutation, Sse, and SLSin the inhibition of neutrophil recruitment by hypervirulent GAS CovS mutants in mouse model of skin infection.
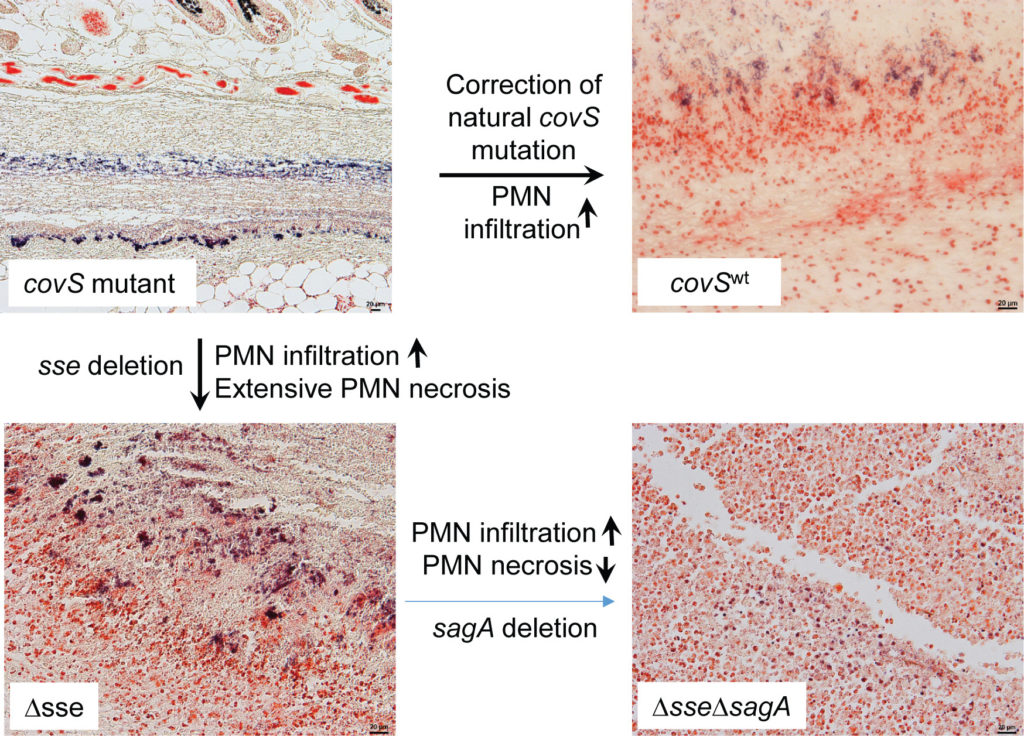
Figure 3.The critical role of CovS mutation and PAF acetylhydrolaseSse and streptolysin S in inhibition of neutrophil recruitment by hypervirulent GAS CovS mutants. In mouse model of subcutaneous infection, hypervirulent GAS CovS mutant severely inhibits neutrophil recruitment, and correction of CovS mutation increases neutrophil recruitment. Deletion of sse increases neutrophils (PMN) recruitment that are largely in necrotic form, and further deletion of sagA, which encodes the peptide component of streptolysin S, leads to neutrophils full of the infection site and reduces neutrophil necrosis. Shown images are microscopic images of infection site sections with Gram stain. GAS and neutrophils were stained in blue and pink colors, respectively. The figure was derived from reference 28.
Vascular Invasion by Hypervirulent GAS CovSMutants
In a mouse model of pulmonary infection, hypervirulent GAS MGAS315 bacteriaare sparsely scattered along the alveolar ducts and alveolar septa right after intratracheal inoculation, and mice are moribund at 24-48 h after inoculation. Alveolar ducts are often severely damaged and contain large numbers of bacteria, bacteria are associated with the septa of alveoli, and, more strikingly, intensive Gram staining for GAS are present in the peribronchovascular and perivascular interstitium (Figure4)[31]. The peribronchovascular interstitium is the connective-tissue sheath that encloses the bronchi, pulmonary arteries, veins, and lymphatic vessels, whereas the perivascular interstitium extended from the peribronchovascular interstitium protects blood vessels that connect the microvasculature system in the alveolar region with the arteries and veins inside the peribronchovascular interstitium. Thus, MGAS315 has the capacity to invade the peribronchovascular interstitium.
MGAS315 has a natural covSG1370T mutation, which results in a CovS G457V missense mutation [9]. This CovS G457Vmutation contributes to the hypervirulence of MGAS315 in subcutaneous infection of mice [9]. An isogenic strain derived from MGAS315 that carries the wild-type covS gene (MGAS315wtcovS) can still cause infections in the alveolar area but cannot invade the peribronchovascular and perivascular interstitium (Figure 4). Deletion of the covS gene in MGAS315wtcovS restored the infection of the peribronchovascular and perivascular interstitium (Figure4). CovS mutation of MGAS315 is required for the peribronchovascular and perivascular invasion.
As MGAS315 invades the perivascular interstitium, the integrity of the smooth muscle and endothelial layers lining the blood vessels became disrupted and separated [31]. As segments of the smooth muscle and endothelial layers are further degraded, the bacteria can enter the lumen of the blood vessels. In contrast, blood vessels are intact in MGAS315wtcovS infection because bacteria infect the alveolar region but do not enter the connective-tissue sheath around blood vessels.MGAS315 loads in the liver and spleen are >1000-fold higher than MGAS315wtcovSin the infections. These data support a critical role of the vascular invasion in systemic MGAS315 dissemination.
The virulence factors that are directly involved in the vascular invasion are not known. Depletion of neutrophils and inflammatory monocytes does not lead to the perivascular invasion in MGAS315wtcovS infection. Thus, the role of CovS mutations in the perivascular invasion appears not to be directly linked to GAS evasion of neutrophil and inflammatory monocyte responses even though the innate immune evasion should be a prerequisite for the perivascular invasion. Hypervirulent GAS may cross the barrier between the alveoli and the peribronchovascular and perivascular interstitium and/or enter the peribronchovascular interstitium by crossing the epithelium in bronchioles. The damages on the smooth muscle and endothelial layers may involve CovRS-controlled virulence factors. Invasion of the vascular system by hypervirulent GAS likely involves coordinated actions of multiple virulence factors that are controlled by CovRS. Elucidation of the invasion route and virulence factors critical for understanding the process of systemic dissemination and developing novel therapies to treat severe invasive GAS infections.
In summary, natural mutations of virulence regulators CovRS, RocA, and RopB enhance GAS virulence. In vivoselection of CovRS mutants requires neutrophils, and CovRS mutants survive better than GAS with wild-type CovRS against neutrophil responses. Downregulation of SpeB due to CovS and RopB mutations contribute to GAS virulence in invasive infections. CovS mutations leads to inhibition of neutrophil recruitment at infection sites, and PAF acetylhydrolaseSse and streptolysin S are both required for and have synergistic contribution to the inhibition of neutrophil recruitment by hypervirulent GAS CovS mutants. CovS mutations also enable GAS to invade the perivascular interstitium and directly attack the vascular system for systemic dissemination. Despite the progress, the molecular mechanisms for in vivo selection of hypervirulent GAS mutants and their vascular invasion remain to be elucidated. Efforts in the elucidation of these mechanisms are expected to provide targets for the prevention and treatment of severe GAS infections.
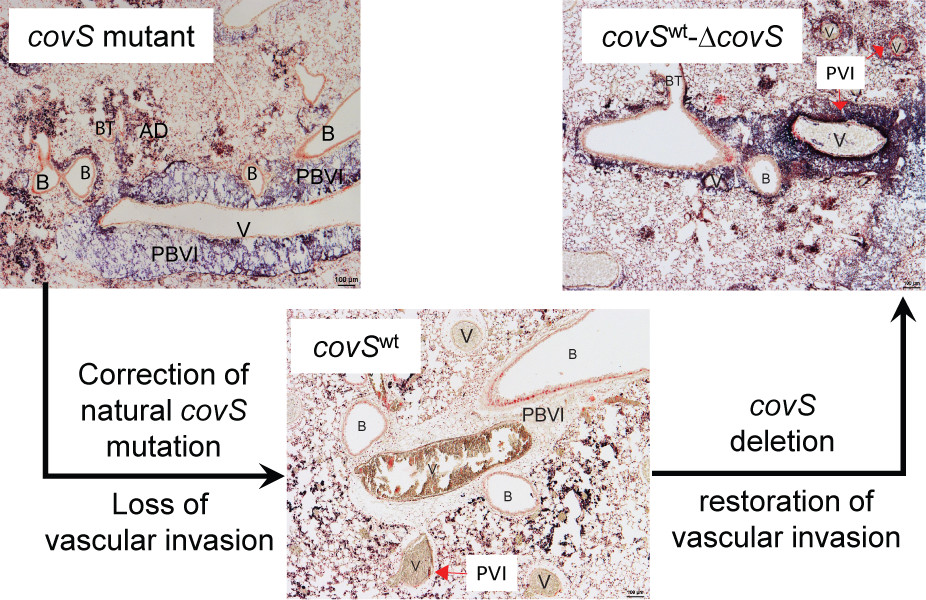
Figure 4.CovS mutations leads to the peribronchovascular and perivascular invasion the vascular attack in murine pulmonary infection. In pulmonary infection of mice from intratracheal GAS inoculation, hypervirulent GAS CovS mutant invades peribronchovascular and vascular invasion, and correction of CovS mutations leads the loss of perivascular and vascular invasions, which are restored by the deletion of the corrected sse gene. Shown are microscopic images of lung sections with Gram stain. Letters: A, alveoli; AS, alveolar septa; AD, alveolar duct; B, bronchiole; BT, bronchial terminus; PBVI, peribronchovascular interstitium; PVI, perivascular interstitium; and V, blood vessel. The figure was derived from reference 31.
Acknowledgements
Acknowledgements
This work was supported in part by grants AI095704 and GM110732 from the National Institutes of Health and the Montana State Agricultural Experimental Station
References
Carapetis JR, Steer AC, Mulholland EK, et al. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005; 5:685–694.
Nelson GE, Pondo T, Toews KA, et al. Epidemiology of invasive group A streptococcal infections in the United States, 2005-2012. Clin Infect Dis. 2016;63:478-486.
Sumby P, Porcella SF, Madrigal AG, et al. Evolutionary origin and emergence of a highly successful clone of serotype M1 group a Streptococcus involved multiple horizontal gene transfer events. J Infect Dis. 2005; 192: 771-782.
Beres SB, Sylva GL, Barbian KD, et al. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. ProcNatlAcadSci USA; 2002; 99: 10078-10083.
Turner CE, Abbott J, Lamagni T, et al. Emergence of a new highly successful acapsular Group A Streptococcus clade of genotype emm89 in the United Kingdom. MBio. 2015; 6: e00622.
Feng W, Liu M, Chen DG, et al. Contemporary Pharyngeal and Invasive emm1 and Invasive emm12 Group A Streptococcus Isolates Exhibit Similar In Vivo Selection for CovRS Mutants in Mice. PLoS One. 2016; 11(9):e0162742.
Sharkawy A, Low DE, Saginur R, et al. Severe group a streptococcal soft-tissue infections in Ontario: 1992-1996. Clin Infect Dis. 2002; 34:454-460.
Li J, Zhu H, Feng W, et al. Regulation of inhibition of neutrophil infiltration by the two-component regulatory system CovRS in subcutaneous murine infection with group A Streptococcus. Infect Immun.2013; 81: 974-983.
Stetzner ZW, Li D, Feng W, et al. Serotype M3 and M28 group A streptococci have distinct capacities to evade neutrophil and TNF-α responses and to invade soft tissues. PLoS One. 2015; 10:e0129417.
Ikebe T, Ato M, Matsumura T, et al. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoSPathog. 2010; 6:e1000832.
Shea PR, Beres SB, Flores AR, et al. Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. ProcNatlAcadSci USA. 2011; 108:5039-5044.
Sumby P, Whitney AR, Graviss EA, et al. Genome-wide analysis of Group A Streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoSPathog. 2006; 2: 41-49.
Engleberg NC, Heath A, Miller A, et al. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis. 2001; 183: 1043-1054.
Kansal RG, Datta V, Aziz RK, et al. Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus J Infect Dis. 2010; 201:855-865.
Lynskey NN, Goulding D, Gierula M, et al. RocA truncation underpins hyper-encapsulation, carriage longevity and transmissibility of serotype M18 group A streptococci. 2013; 9:e1003842.
Miller EW, Danger JL, Ramalinga AB, et al. Regulatory rewiring confers serotype-specific hyper-virulence in the human pathogen group A Streptococcus. 2015; 98:473-489.
Walker MJ, Hollands A, Sanderson-Smith ML, et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med. 2007; 13: 981-985.
Liu G, Feng W, Li D, et al. The MgaRegulon but Not Deoxyribonuclease Sda1 of Invasive M1T1 Group A Streptococcus Contributes to In Vivo Selection of CovRS Mutations and Resistance to Innate Immune Killing Mechanisms. Infect Immun. 2015; 83: 4293-4303.
Cole JN, Pence MA, von Köckritz-Blickwede M, et al. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. 2010; 1: e00191-10.
Li J, Liu G, Feng W, et al. Neutrophils select hypervirulentCovRS mutants of M1T1 group A Streptococcusduring subcutaneous infection of mice. Infect Immun. 2014; 82:1579-1590.
Feng W, Minor D, Liu M, et al. Null Mutations of Group A Streptococcus Orphan Kinase RocA: Selection in Mouse Infection and Comparison with CovS Mutations in Alteration of In Vitro and In Vivo Protease SpeB Expression and Virulence. Infect Immun. 2017; 85:e00790-16.
Aziz RK, Pabst MJ, Jeng A, et al. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. MolMicrobiol. 2004; 51:123-134.
Dalton TL, Hobb RI, Scott JR. Analysis of the role of CovR and CovS in the dissemination of Streptococcus pyogenes in invasive skin disease. MicrobPathog. 2006; 40(5):221-7.
Engleberg NC, Heath A, Vardaman K, et al. Contribution of CsrR-regulated virulence factors to the progress and outcome of murine skin infections by Streptococcus pyogenes. Infect Immun. 2004; 72:623-628.
Olsen RJ, Raghuram A, Cantu C, et al. The majority of 9,729 group A streptococcus strains causing disease secrete SpeB cysteine protease: pathogenesis implications. Infect Immun. 2015; 83:4750-4758.
Bakleh M, Wold LE, Mandrekar JN, et al. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005; 40:410-414.
Liu M, Zhu H, Li J, et al. Group A Streptococcus secreted esterase hydrolyzes platelet-activating factor to impede neutrophil recruitment and facilitate innate immune evasion. PLoSPathog. 2012; 8:e1002624.
Feng W, Minor D, Liu M, et al. Requirement and Synergistic Contribution of Platelet-Activating Factor AcetylhydrolaseSse and Streptolysin S to Inhibition of Neutrophil Recruitment and Systemic Infection by Hypervirulent emm3 Group A Streptococcus in Subcutaneous Infection of Mice. Infect Immun.2017; 85: e00530-17.
Lin A, Loughman JA, Zinselmeyer BH, et al. Streptolysin S inhibits neutrophil recruitment during the early stages of Streptococcus pyogenes infection. Infect Immun. 2009; 77:5190-5201.
Miyoshi-Akiyama T, Takamatsu D, Koyanagi M, et al. Cytocidal effect of Streptococcus pyogenes on mouse neutrophils in vivo and the critical role of streptolysin S. J Infect Dis. 2005; 192:107-116.
Lei B, Minor D, Feng W, et al. Hypervirulent Group A Streptococcus of Genotype emm3 Invades the Vascular System in Pulmonary Infection of Mice. Infect Immun. 2018; 86: e00080-18.
Received: July 15, 2018;
Accepted: August 15, 2018;
Published: August 17, 2018
To cite this article : Mengyao Liu, Lei B. Pathogenesis of Hypervirulent Group a Streptococcus. Japan Journal of Medicine. 2018: 1:6.
©Liu M, et al. 2018.