
Research Article / Open Access
DOI: 10.31488/jjm.1000141
Proven or Highly Probable Factor VII Arg304Gln (FVII Padua) Homozygous Defect: A Mutation that Occurred in Different Areas of the World Which Were Related or Unrelated With Known Forced or Spontaneous Migratory Patterns
Antonio Girolami1, Silvia Ferrari1, Elisabetta Cosi1, Claudia Santarossa1, Emanuel Sueldo2, Mirta Arias2
Dept. of Medicine, University of Padua Medical School, Padua, Italy
Laboratorio de Hematologia y Hemostasia, Unidad Asistencial Dr. Cesar Milstein, Buenos Aires, Argentina
*Corresponding author:Prof. A. Girolami, D.P.M.S., Department of Medicine, Via Ospedale 105, Padua, 35128, Italy, Tel: 0039-049-8213026; Fax: 0039-049-657391;
Abstract
Objective: to investigate the areas in which the Factor VII Padua Arg304Gln mutation has been reported and to identify its diffusion according to known migratory patterns. Patients, Methods: countries that showed the presence of at least 2 unrelated proven homozygotes and six heterozygotes were selected. These patients had to be studied by molecular biology techniques (Group A). Patients suspected to be cases of FVII Padua only on the basis of clotting tests were also gathered (Group B). The latter patients had to show the discrepancy in FVII assays between rabbit brain (low activity) vs human recombinant and Ox brain thromboplastins (normal activity). Results: there are 5 areas of the world that met the criteria, namely Mediterranean area, Iran, USA, Japan and Argentina. Some of these countries can be related because of ancient migratory patterns. Conclusions: It is likely that at least two independent main sources for the Arg304Gln Factor VII mutation, have occurred, namely the Mediterranean area and Japan pattern. The observation that several heterozygotes for this mutation have been found in West Africa suggests that the homozygotes seen in the USA only among African-Americans are the result of the “forced” emigration that occurred in the past.
Keywords: factor VII deficiency, FVII Padua, migrations, bleeding, diffusion
Introduction
Congenital FVII deficiency is the most frequent condition among the Rare Bleeding Disorders. Its prevalence is estimated to be around 1:500.000 persons [1]. The defect has been first reported in the USA in 1955 [2] and subsequently all over the world. About 200 mutations, mainly missense ones, have been described. Factor VII Padua was first reported in Northeastern Italy in 1978 [3]. The Arg304Gln mutation in Exon 8 responsible for the defect was independently seen in a single patient in England and in several patients in Northeastern Italy (Padua) both in 1991 [4,5]. Subsequently, several other patients were seen in several Mediterranean countries (France, Tunisia, Israel), in Japan, in Iran and in the USA (6-19). All cases seen in the USA were described among the African-American population [13,17,18].
Sporadic cases were also seen in other countries [4,20] but the main areas remained the four sites cited above. Recently, two homozygous patients were reported in Latin America (Argentina) [21,22]. These investigations seem to suggest that the mutation has a multiple origin in at least five areas of the world, namely Italy (Mediterranean area), Japan, Iran, USA and Argentina. The purpose of the present study was to investigate the significance of the same mutation occurring in different continents and to attempt and explain the findings on the basis of known “forced” or spontaneous migratory patterns which occurred in the past.
Patients and Methods
Proven or probable homozygotes for the Factor VII Arg304Gln mutation were gathered from two sources namely: 1) files of personal patients investigated in the past and 2) two time-unlimited Pub Med Searches carried out in June 2016 and June 2018. The patients who were investigated studied in Padua were informed and gave their consent to the study which was carried out according to the Helsinki Convention.
All cases investigated by molecular biology techniques were directly included providing that each country or area had at least two unrelated homozygotes and at least five heterozygotes. These patients were considered as proven cases of FVII Padua defect (Group A). Compound heterozygotes containing the Arg304Gln mutation were recorded but were disregarded as inclusion criteria. Cases without molecular biology techniques (Group B) were included if they met the following criteria: 1) low FVII levels using rabbit brain thromboplastin (less than 10% of normal) 2) normal FVII level using ox brain thromboplastin (100% of normal), 3) intermediate FVII levels using human placenta thromboplastin or genetically produced human thromboplastin (50-70% of normal), 4) global test such as PT and or Thrombotest should have the same behavior, 5) correction of the defect by the addition of normal plasma or serum. Factor VII antigen was not considered as an inclusion criterium but it was recorded whenever available. The geographical distribution of the homozygous patients investigated were tentatively compared with known “forced” or spontaneous migratory patterns in an attempt to establish a potential relation among the patients found in the different areas of the world.
Results
The evaluation of the distribution of patients with this abnormality suggests a prevalent concentration in the following geographical regions: Italy (Mediterranean area), Japan, USA, Iran and Argentina. The number of sure homozygotes (Group A) in these areas were 17,2,3,6 and 2, respectively for a total of 30 cases. That of probable homozygotes (Group B) was instead 3,1,17, zero and 2, respectively for a total of 23 patients (Table 1).
Table 1. Proven and probable homozygous patients with the Arg304Gln (FVII Padua) mutation in the world
| Country | Proven homozygotes | Probable homozygotes | Comments |
|---|---|---|---|
| Italy and other Mediterranean countries
Japan USA Iran Argentina | 17 2 3 6 2 |
3 1 17 None? 2 |
Mediterranean countries (Tunisia, Spain, France, Israel) All patients are African-Americans |
The cases in the Mediterranean area were concentrated mainly in Italy but cases were also reported from France, Tunisia and Israel [8-12]. In no other area of the world there is a similar prevalence of the mutation. The cases from Japan apparently come from different islands that compose the country [16,19]. Those from the USA come mainly from Southern States and involve always African Americans [13,17,18]. The patients from Iran appear to come from several areas of the country [14,15]. The homozygous cases from Latin America appear to be concentrated in the Northern part of Argentina. Several heterozygotes have been described in Brazil, Venezuela and Costa Rica [21,22]. Compound heterozygotes have been reported from China, USA France and North Africa (Table 2) [8,9,23-28].
Table 2. Compound heterozygotes involving the Arg304Gln mutation. n.r.= not reported.
| Authors (year) | Age/gender | FVII activity Rabbit, Human, OX | FVII Antigen (%) | Bleeding | Associated mutation | Country | Comments |
|---|---|---|---|---|---|---|---|
|
Giamsy-Blaizot et al (2001) |
1) n.r., M
2) n.r., F
3) n.r.M
|
5, n.r., n.r.
<5, n.r., n.r.
3, n.r., n.r.
|
54
55
23 |
n.r.
n.r.
n.r.
|
Ala244Val
Cys135Arg
Gly180Arg |
North Africa
France
France |
Country unspecified ; Different kindreds |
| Ding et al.
(2003) |
n.r., n.r. | n.r., n.r., n.r. | n.r. | Mild | Arg304Trp | China | |
| Pollak et al.
(2006) |
27, F | 26 54 n.r. | 67 | none | Arg315Trp | USA | African-American |
| Marty et al.
(2007) |
57, F | 10 n.r. n.r. | 73 | Mild
(easy bruising) |
Cys135Arg | France | Patient had also a post surgery DVT |
| Hermann et al.
(2009) |
5, F | 6 n.r. n.r. | n.r. | Moderate (gastro-intestinal) | Gly365Cys | USA | |
| Jin et al.
(2015) |
n.r., M | 2 n.r. n.r. | n.r. | Mild | His348Gln | China | |
| Kuppurswamy et al (1993) | n.r., n.r. | 4, n.r., n.r. | n.r. | none | Gln100Arg | USA | FVII Kansas |
Discussion
It is difficult to envisage a common origin for all these five areas of the world. It is likely that only the patients seen in Japan represent a true independent source. This is consistent with the isolation in which the country lived for centuries (Figure 1). On the contrary, a plausible link may exist between the Mediterranean area (Italy, Spain, Israel, Tunisia) and Latin-America (L-A). In this regard it is worth noting that a few heterozygotes but no homozygotes, with this mutation had been reported from three L-A countries, Costa Rica, Venezuela and Brazil [23,24].
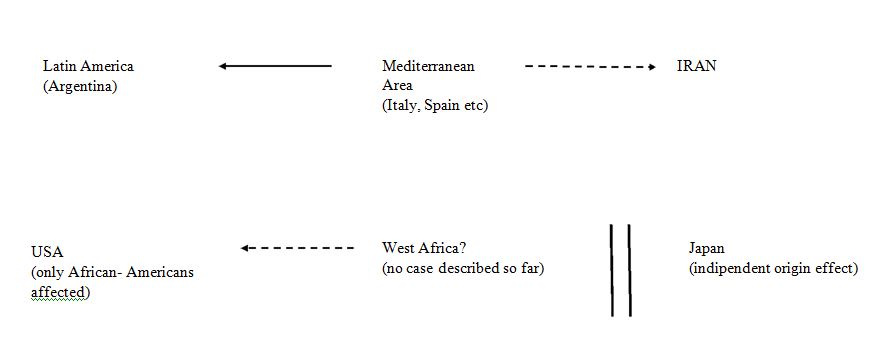
Figure 1.Schematic interpretation of the role played by migratory routes. Broken arrows indicate possible routes; solid arrows indicate proven or likely routes.
The cases seen in Iran are probably also the result of migratory contacts with the Mediterranean area. Contacts between Mediterranean countries (Morocco, Israel) and Middle East countries (Iran) have already been postulated for other FVII defects [29]. The relation between the Mediterranean area and Latin America is easily understandable. The main waves of spontaneous emigration to that continent from European countries are three, namely 1) The colonization period (1500-1800), Spain and Portugal contributed most of the people during these centuries, 2) The years between 1850 and 1915 which involved mainly Italy and Spain, 3) the post Second World War period. The European countries involved in this wave of emigration were again Italy and Spain but also included Germany and other European countries.
No proven FVII Padua case has been reported from Spain but there are a few probable cases as indicated by clotting tests [6,30]. Furthermore, one must note that the Northeastern part of Italy where FVII Padua was first reported has supplied a large fraction of the second wave of emigrants to Argentina [3,30]. Due to the lack of any ethnic relation between European immigration and the African American patients seen in the USA it is conceivable that an independent origin might have occurred [13,31]. In this regard it is worth noting that the index patient reported by Alexander et al. was instead a Caucasian and had a severe FVII deficiency [2].
The majority of African Americans derive from slave operations carried out by slave traders who forcibly transported captured persons from West Africa to the USA or, to a minor extent, to Brazil and to central Latin America countries during three centuries (1550-1850) [32,33]. As a consequence, the Arg304Gln mutation should be found among the population of West African countries such as Senegal, Gambia, Liberia, Ghana, Nigeria and others.
Unfortunately, no homozygous case with this defect has been reported from this area of Africa. The only available paper on the subject fails to report patients with FVII deficiency that could be suspicious of being a case of FVII Padua [34]. 49 African-Brazilians were investigated for the presence of the Arg304Gln mutation but none was found [24].
The problem should be clarified only by means of an extensive investigation of the West Africa populations. Important data in this regard have been supplied recently by the “1000 Genomes Project Consortium investigation [35,36]”. These studies have shown the presence of 12 heterozygotes for the FVII Arg304Gln mutation among 661 healthy African subjects (1,8%) and 3 heterozygotes among 61 African-American healthy subjects of the southwest USA (2,5%).
Furthermore the mutation was not found among other ethnic groups of the same area of the USA. Taken altogether these observations support the African origin of the Arg304Gln mutation found in the USA. Compound heterozygotes have been reported in several countries (Table 2), confirming that the Arg304 residue is a hot spot for mutations [6, 7]. The strict association between the clotting abnormalities and the Arg304Gln mutation has been repeatedly confirmed [13,17,18,37]. However, it has to be remembered that other mutations may yield similar discrepancies in FVII activity assays. For example, FVII Nagoya (Arg304Trp) yields a clotting pattern similar to that of FVII Padua [38]. However, the Arg304Trp mutation is rare, having been described mainly in Japan, and therefore the peculiar clotting pattern discrepancies in FVII activity assays almost surely indicate the existence of the Arg304Gln defect [39].
This indicates that the residue Arg 304 plays a fundamental role in the interaction with Tissue Factor (T.F.). It has to be noted that, besides the catalytic domain of exon 8, another area of the FVII molecule is involved in interaction with T.F. This is the area of the first Endothelial Growth Factor (EGF) controlled by exon 4. However, the defects in this area are also rare and, furthermore, the pattern of activation is not as typical as that presented by FVII Padua [40,41]. The correct diagnosis of the Arg304Gln mutation has important clinical implications.
Since these patients show a mild bleeding tendency or are asymptomatic [3,6,13], one has to be very careful with the replacement therapy to avoid possible thrombotic complications [42-44]. Since the Arg304Gln mutation is rare in Northern Europa (45,46) it may be stated that the main sites are represented by the Mediterranean area, West Africa and the USA as a result of “forced” emigration from West African countries. The last conclusion is supported by the observation that all true homozygotes seen in the USA are African Americans [43,44].
Acknowledgements
The study was performed according to the Helsinki Convention. Patients studied in Padua were duly informed and agreed with the study. This study was supported in part (secretarial assistance) by the “Associazione Emofilia ed altre Coagulopatie delle Tre Venezie”.
Conflict of Interest
The Authors declare that they have no conflict of interest.
References
Perry D. Factor VII deficiency. Br J Haematol. 2002; 118: 689–700.
Alexander B, Goldstein R, Landwehr G, et al. Congenital spca deficiency: a hitherto unrecognized coagulation defect with hemorrhage rectified by serum and serum fractions. J Clin Invest. 1951; 30: 596–608.
Girolami A, Fabris F, Dal Bo Zanon R, et al. Factor VII Padua: a congenital coagulation disorder due to an abnormal factor VII with a peculiar activation pattern. J Lab Clin Med. 1978;91:387–395.
O’Brien DP, Gale KM, Anderson JS, et al. Purification and characterization of factor VII 304-Gln: a variant molecule with reduced activity isolated from a clinically unaffected male. Blood. 1991;78:132–140.
James HL, Kumar A, Girolami A, et al. Variant coagulation Factor X and VII with point mutations in a highly conserved motif in the substrate binding pocket, comparative molecular modelling. Thromb Haemost. 1991;69(Suppl.):937 (Abstract).
Girolami A, Berti de Marinis G, Bonamigo E, et al. Worldwide diffusion of FVII Arg304Gln coagulation defect (FVII Padua). Eur J Haematol. 2011;86:135-139.
Bernardi F, Liney DL, Patracchini P, et al. Molecular defects in CRM+ factor VII deficiencies: modelling of missense mutations in the catalytic domain of FVII. Br J Haematol. 1994;86:610–618.
Marty S, Barro C, Chatelain B, et al. The paradoxical association between inherited factor VII deficiency and venous thrombosis. Haemophilia. 2008;14:564–570.
Giansily-Blaizot M, Aguilar-Martinez P, Biron-Andreani C, et al. Analysis of the genotypes and phenotypes of 37 unrelated patients with nherited factor VII deficiency. Eur J Hum Genet. 2001;9:105–12.
Horellou MH, Chalendard J, Juin F, et al. Source of tissue factor leading to discrepant plasma FVII levels in eight FVII deficient patients of African origin. J Thromb Haemost. 2007;5(Suppl. 2):P-M-002
Fromovich-Amit Y, Zivelin A, Rosenberg N, et al. Characterization of mutations causing factor VII deficiency in 61 unrelated Israeli patients. J Thromb Haemost. 2004;2:1774–1781.
Fromovich-Amit Y, Zivelin A, Rosenberg N, et al. Of four mutations in the factor VII gene in Tunisian patients, one novel mutation (Ser339Phe) in three unrelated families abrogates factor X activation. Blood Coag Fibrinolysis. 2005:16:369-374.
Kirkel D, Lin TW, Fu SW, et al. Asymptomatic factor VII deficiency: gene analysis and structure-function relationships. Blood Coagul Fibrinolysis. 2010;21:91–94.
Peyvandi F, Jenkins PV, Mannucci PM, et al. Molecular characterization and three-dimensional structural analysis of mutations in 21 unrelated families with inherited factor VII deficiency. Thromb Haemost. 2000;84:250–257.
Tahatahaly A, Ravanbod S, Baghapour M, et al. Factor VII genotype and clinical profile in 6 Iranian patients with congenital FVII deficiency. J Thromb Haemost. 2007;5(Suppl.):1405 (Abstract).
Sekiya A, Morishita E, Maruyama K, et al. Significant decrease in factor VII activity by tissue thromboplastin derived from rabbit brain in a patient with congenital factor VII deficiency (FVII Padua). Rinsho Ketsueki. 2012;53:357-560.
Sabharwal AK, Kuppuswamy MN, Foster DC, et al. Factor VII deficiency (FVII Richmond , R304Q Mutant) associated with thrombosis. Circulation. 1992;86(Suppl.):679 (Abstract).
Shurafa MS, Kumar A, Fair DS, et al. The molecular defect in factor VII Detroit is due to substitution of Arg (304) by Glu. Faseb J. 1993;7:115 (Abstract).
Takamiya O, McVey I, Kemhell-Cook G, et al. Dysfunctional human FVII. Detection of missense mutations by PCR and single-strand conformational variants of coagulation factor VII. Hum Molec Genet.1993;2:1355-1359.
Vanrusselt A, Vermylen J. Congenital FVII dysfunction with a different activities towards human, bovine or rabbit brain thromboplastins. Thromb. Hemost. 1993;69:1293 (Abstract)
Girolami A, Arias M, Sueldo E, et al. First report of homozygous factor VII Padua (Arg304Gln) defect in a family from Argentina. Hematology & Medical Oncology 2016; 1:1-5.
Girolami A, Sueldo E, Ferrari S, et al. Report of the second patients with a homozygous Factor VII Padua (Arg304Gln) defect in northern Argentina. Mathews J Case Rep. 2018; ISNN 2474-36666.
Herrmann FH, Wulff K, Auerswald G, et al. Factor VII deficiency: clinical manifestation of 717 subjects from Europe and Latin America with mutations in the factor 7 gene. Haemophilia. 2009;15:267-280.
Rodrigues DN, Siqueira LH, Galizoni AM, et al. Prevalence of factor VII deficiency and molecular characterization of the F7 gene in Brazilian patients. Blood Coagul Fibrinolysis. 2003;14:289-292.
Ding QL, Wang HL, Wang XF, et al. [Inherited coagulation factor VII deficiency caused by double heterozygotic mutations Arg304Gln and Arg304Trp]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2003;20:279-283.
Wang Y, Hao X, Yang L, et al. Genetic analysis of a pedigree with hereditary coagulation factor VII deficiency. Chinese Journal of Medical Genetics.2015; 32:222-225.
Kuppuswamy M, Sahbarwal A, Birktoff J, et al. Molecular characterization of human factor-VII Kansas (gk704)- substitution of Gln100 by Arg in one allele and of Arg304 by Gln possibly in the other allele. Thromb. Haemost. 1993;69:1291 (Abstract).
Pollak ES, Russell TT, Ptashkin B, et al. Asymptomatic factor VII deficiency in African Americans. Am J Clin Pathol. 2006;126:128-132.
Tamary H, Fromovich Y, Shalmon L, et al. Ala244Val is a common, probably ancient mutation causing factor VII deficiency in Moroccan and Iranian Jews. Thromb Haemost. 1996;76:283-291.
Pardo A, Oteyza JP, Blanco L, et al. Study of Different Factor VII Deficiency Variants in Nine Families from Spain. Haemostasis. 1987;17:268–272.
Girolami A, Bonamigo E, Vettore S. The lack of ties between northeastern Italy and Afro-American suggest a multifounder effect for FVII Padua (Arg304Gln) disorder. Blood Coag Fibrin. 2010;21:775-776.
World Book Encyclopedia: Slavery; vol 17; pag 415, Negro, vol 14, pag 106. Field Educational Corporation, Chicago. 1972.
Encyclopedia Britannica: Slavery- William Berstant Pub. Chicago, 1963;20:703-782.
Dokekias AE, Bopaka R, Malanda F, et al. Evaluacion des desordres ongenitaux de l’hemostase en el Hopital Universitaire de Brazzaville, Congo. Med Trop 2009;69:102–103.
About 1000 Genomes http://www.internationalgenome.org/about
Auton A, Abecasis G. A global reference for human genetic variation. The 1000 Genomes Project Consortium. Nature. 2015; 526:68-87.
James H, Girolami A, Hubbard J, et al. The dysfunction of coagulation factor VIIPadua results from substitution of arginine-304 by glutamine. Bioch. Biophys Acta. 1993; 1172: 301-305.
Matsushita T, Kojima T, Emi N, et al. Impaired human tissue factor-mediated activity in blood clotting factor VII Nagoya (Arg304 fi Trp). Evidence that a region in the catalytic domain of factor VII is important for the association with tissue factor. J Biol Chem. 1994;269:7355–7363.
Girolami A, Berti de Marinis G, Bonamigo E, et al. Ox brain versus rabbit brain thromboplastin assays are the best tool for a preliminary diagnosis of the Arg304Gln factor VII defect (FVII Padua). Acta Haematol. 2010;124:229-234.
Clarke BJ, Ofosu FA, Sridhara S, et al. The first epidermal growth factor domain of human coagulation factor VII is essential for binding with tissue factor. FEBS Lett. 1992; 298:206-10.
Dickinson CD, Kelly CR, Ruf W. Identification of surface residues mediating tissue factor binding and catalytic function of the serine protease factor VIIa. Proc Natl Acad Sci U S A. 1996;93:14379-84.
Girolami A, Bertozzi I, Rigoni I, et al. Congenital FVII deficiency and thrombotic events after replacement therapy. J Thromb Thrombolysis. 2011;32:362-367.
Triplett DA, Brandt JT, Batard MA, et al. Hereditary factor VII deficiency: heterogeneity defined by combined functional and immunochemical analysis. Blood. 1985;66:1284-1287.
Barnett JM, Demel KC, Mega AE, et al. Lack of bleeding in patients with severe factor VII deficiency. Am J Hematol. 2005;78:134-137.
Cooper DN, Millar DS, Wacey A, et al. Inherited factor VII deficiency: molecular genetics and pathophysiology. Thromb Haemost. 1997;78:151–160.
Millar DS, Kemball-Cook G, McVey JH. Molecular analysis of the genotype–phenotype relationship in factor VII deficiency. Hum Genet. 2000;107:327–342.
Received: April 10 2019;
Accepted: May 20, 2019;
Published: May 23, 2019.
To cite this article : Girolami A, Ferrari S, Cosi E, et al. Proven or Highly Probable Factor VII Arg304Gln (FVII Padua) Homozygous Defect: A Mutation that Occurred in Different Areas of the World Which Were Related or Unrelated With Known Forced or Spontaneous Migratory Patterns. Japan Journal of Medicine. 2019: 2:3.
© Girolami A, et al. 2019.